 |
Revista Digital Universitaria |
| ISSN: 1607 - 6079 Publicación mensual | |
| 1 de marzo de 2010 Vol.11, No.3 |
| EJEMPLARES | DIRECTORIO | QUIÉNES SOMOS | COLABORA | VÍNCULOS | MAPA DE SITIO |
|
||||||
|
||||||
|
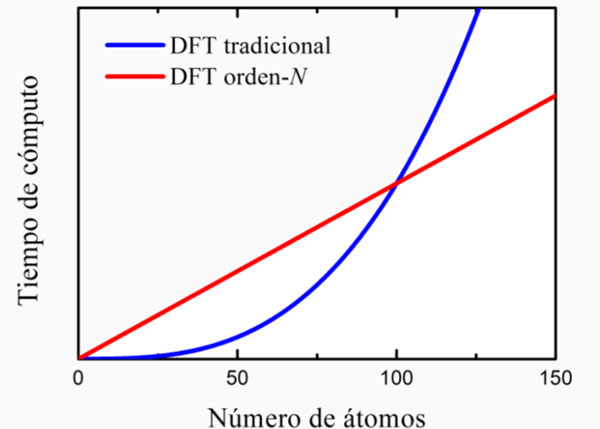
Diseño asistido por computadora El diseño de nuevos materiales a partir de fundamentos puramente teóricos o ab-initio ha sido el sueño de generaciones de científicos. Dado que, en principio, cada una de las propiedades de todos los materiales pueden describirse por la mecánica cuántica, uno podría reemplazar el método de prueba y error por el diseño computacional a primeros principios de materiales y dispositivos. Sin embargo, existen numerosos problemas técnicos que impiden este sueño. Primeramente, el problema cuántico de muchas partículas fuertemente correlacionadas sigue siendo un reto para los físicos. Los métodos numéricos, tales como la interacción de configuraciones o el Monte Carlo cuántico, son capaces -en principio- de resolver con precisión la ecuación de Schrödinger de muchos cuerpos. Sin embargo, debido a su extremadamente alto costo computacional, el uso de estos métodos es muy limitado en la práctica, por lo que en la mayoría de los casos se utiliza la aproximación de campo medio, en la que se aborda únicamente la dinámica de una partícula en presencia de una interacción promedio con las demás partículas, tales como en los métodos de Hartree-Fock o funcional de la densidad. En esta última se introduce una energía de correlación e intercambio que le permite incluir algunos efectos más allá de campo medio. Aún cuando el problema es reducido al de una sola partícula, se tienen que diagonalizar matrices del tamaño de los grados de libertad del sistema, si la función de onda de dicha partícula es expresada en una base de orbitales atómicos. Por ejemplo, en una molécula de hidrógeno ionizada se tiene un electrón interactuando con dos núcleos y el problema cuántico del electrón puede escribirse como una matriz de 2x2. En general, dentro de la aproximación de campo medio, el tamaño de la matriz es al menos igual al número de átomos del sistema, debido a la naturaleza multi-orbital de los átomos. Aunado a esta complicación relacionada al tamaño de matriz, las ecuaciones de campo medio deben resolverse de forma autoconsistente, es decir, partiendo de una configuración arbitraria de partículas se calcula el potencial promedio y se obtiene una solución de las ecuaciones cuánticas que conduce a una nueva configuración que sirve como punto de partida para repetir el proceso hasta lograr autoconsistencia. Por otro lado, se sabe que el tiempo computacional del proceso de diagonalización crece de forma cúbica con el tamaño de la matriz en ausencia de simetría, por lo que el cómputo actual es capaz de abordar únicamente sistemas menores que 106 átomos. En 1965 Gordon E. Moore, cofundador de Intel, propuso una ley empírica conocida como la ley de Moore, en la que se estima que la densidad de transistores en un circuito integrado se duplica cada dos años; en consecuencia, la capacidad de cómputo se duplica en el mismo periodo. Esta ley se ha cumplido durante las últimas cuatro décadas. Si la ley de Moore siguiera cumpliéndose aún para los transistores subatómicos, se requerirían siglos para poder abordar sistemas macroscópicos arbitrarios partiendo de la mecánica cuántica a primeros principios. Uno de los métodos cuánticos más utilizados en la actualidad se basa en la teoría del funcional de la densidad (DFT, por sus siglas en inglés), por la que Walter Kohn recibiera el premio Nobel de química en 1998. En dicha teoría se supone que la cantidad básica de la mecánica cuántica podría ser la densidad y no la función de onda, teniendo la virtud de ser más fácil y rápida de calcular que la función de onda al no importar la fase de la misma. En particular, la solución de las ecuaciones de Kohn-Sham requiere un tiempo de cómputo que se incremente de forma cúbica con el número de átomos, similar a los demás métodos de campo medio. En los últimos años, se está desarrollando una técnica llamada escalamiento lineal u orden-N con el fin de mejorar la eficiencia de DFT. 2 Dicha técnica se resume en la frase “divide y vencerás”, ya que se divide al sistema en pequeños subsistemas a causa de la interacción coulombiana, que es fuertemente apantallada en un sistema de partículas cargadas, siendo esta interacción la más importante en la escala atómica. En consecuencia, se necesita considerar únicamente la interacción con las partículas más cercanas. Por ejemplo, la diagonalización de una matriz de 1000x1000 puede llevarse a cabo realizando 1000 diagonalizaciones de matrices de 10x10. El tamaño de estas últimas depende del número de vecinos considerados. Por lo que el tiempo de cómputo es proporcional al tamaño del sistema. En la figura 2 se muestra la ventaja del método de escalamiento lineal para sistemas mayores que 100 átomos en comparación con los métodos tradicionales. Hoy en día, mediante DFT tradicional se pueden tratar únicamente cientos de átomos y con el escalamiento lineal esta cifra alcanza millones de átomos. Desafortunadamente, aún usando este último método se tiene que esperar del orden de un siglo para poder abordar sistemas de 1023 átomos en caso de que la ley de Moore se siguiera cumpliendo.
Existen otras estrategias para resolver el problema de tamaño. Se ha propuesto conectar secuencialmente los métodos de diferentes escalas, es decir, se usan los resultados de un cálculo de menor escala como parámetros de otro de mayor escala, comenzando con cálculos de primeros principios y concluyendo en predicciones macroscópicas. Además de este tipo de métodos, para sistemas con simetrías particulares existen otros, específicos, altamente eficientes. En el caso de sólidos cristalinos, donde los átomos se ordenan periódicamente, se utiliza el concepto de espacio recíproco, cuya unidad es el inverso de la distancia en cada dirección, de tal forma que los eventos a escala macroscópica ocurren en la zona central reducida del espacio recíproco, llamada la primera zona de Brillouin.3 En este método se supone que la extensión de los sólidos es infinita, su estructura perfecta, y sus propiedades se obtienen realizando integrales sobre dicha zona. Cabe mencionar que la predicción de Shockley del transistor se fundamentó en la teoría de bandas expresada en el espacio recíproco. Para el caso de sólidos con desorden estructural de largo alcance, o bien con presencia de defectos, impurezas o superficies, se requieren métodos basados en el espacio real. Uno de los más eficientes utiliza la idea de renormalización propuesta originalmente por Leo Kadanoff. En particular, dicha renormalización aplicada al estudio de los cuasicristales ha permitido abordar sólidos aperiódicos de tamaño macroscópico incluyendo defectos e impurezas, dado que su tiempo de cómputo crece en forma logarítmica con el tamaño del sistema.4 Para dar una idea de su eficiencia, un sistema de 1023 átomos tardaría únicamente 23 veces más que uno de 10 átomos. Como se puede apreciar, aún estamos lejos del sueño del diseño de materiales partiendo únicamente de los primeros principios. Sin embargo, existen algunos materiales en los que la predicción de sus propiedades mediante la capacidad de cómputo actual ha sido muy exitosa, como los materiales catalíticos, las moléculas farmacológicas, los nanomateriales, entre otros. A continuación comentaremos en detalle algunos de ellos. 2. S. Goedecker. "Linear
scaling electronic structure methods",
Rev. Mod. Phys. (1999) 71, 1085.
|
||||||
| D.R. © Dirección General de Servicios de Cómputo Académico-UNAM / Ciudad Universitaria, México D.F. Se autoriza la reproducción total o parcial de los artículos aquí presentados, siempre y cuando se cite la fuente completa y su dirección electrónica |
||||||



 |
E
d i t o r i a l |
|
Perspectivas del Supercómputo en México |
El impacto del Supercómputo en la química cuántica
|
|
Supercómputo en la física de la materia condensada:dos ejemplos
|
||
Materiales por diseño ¿ficción o realidad?
|
||
El
Supercómputo en 2010
|
||
| INNOVACIÓN EN TICS | ||
| DIVULMEDIA | PRESENCIA UNIVERSITARIA | |
 |
 |