

Revista Digital Universitaria ISSN: 1607 - 6079 | Publicación mensual
1 de junio de 2014 vol.15, No.6
• TEMA DEL MES •
Medicina personalizada
Introducción
El cáncer es una enfermedad del genoma. Esta noción empezó a tomar forma desde 1890, cuando el patólogo alemán David Von Hansemann, a través de un análisis detallado de 13 diferentes tipos de tumores diferentes, postuló la idea de que el cáncer estaba asociado con alteraciones en la segregación de los cromosomas durante la división celular. Posteriormente Theodor Bovery, mediante la manipulación experimental de huevos de erizo de mar, descubrió que una “combinación incorrecta de cromosomas” era capaz de generar células con una capacidad de crecimiento ilimitado. Gracias a sus observaciones, Bovery fue incluso capaz de prever la existencia de conceptos fundamentales para la biología del cáncer, tales como los puntos de control del ciclo celular, la existencia de genes supresores y |
En este mismo periodo se iniciaron las discusiones acerca de la factibilidad de llevar a cabo el Proyecto del Genoma Humano (PGH) con la finalidad de conocer el orden exacto de las más de 3,200 millones de bases del DNA del ser humano. | |
 |
||
En años posteriores, esta visión fue haciéndose más sólida gracias a diversos hallazgos y descubrimientos, tales como la identificación de virus capaces de provocar cáncer (el virus del sarcoma de Rous, descubierto por Peyton Rous, en 1910), la identificación de la primera alteración cromosómica específica en un tumor (el cromosoma Filadelfia, identificado en 1960 en pacientes con leucemia mieloide crónica), la descripción del papel de los genes supresores de tumor (principalmente RB1 y TP53 en la década de los setentas y ochentas), el descubrimiento de homólogos humanos de oncogenes virales (SRC, en 1976, por el grupo de Harold Varmus, Michael Bishop y Deborah Spector) y la clonación del primer oncogén humano (1982, HRAS, por los grupos de Weinberg, Wigler y Barbacid).
De esta forma, para finales de los años ochentas ya se había identificado la mayoría de los mecanismos mutagénicos existentes en cáncer y se había identificado una variedad de oncogenes activados por cada uno de estos mecanismos (mutaciones puntuales, KRAS en cáncer de colon; translocaciones cromosómicas BCR-ABL1 en leucemias; pérdida, deleciones de RB1 en retinoblastoma o ganancia de material genético; amplificaciones de MYC en cáncer de pulmón).
El Proyecto del Genoma Humano y la investigación en cáncer
En este mismo periodo se iniciaron las discusiones acerca de la factibilidad de llevar a cabo el Proyecto del Genoma Humano (PGH) con la finalidad de conocer el orden exacto de las más de 3,200 millones de bases del DNA del ser humano.El PGH fue blanco de diversas y severas críticas sobre todo en cuanto a su factibilidad, debido a los altos costos y a la baja eficiencia de los métodos de secuenciación de DNA existentes en los primeros años de los ochentas, cuando la mayoría de los genomas secuenciados eran de diferentes virus cuyo tamaño no era mayor a los 2,000 pares de bases.
Uno de las primeros trabajos que apoyaron de forma abierta al PGH, discutiendo los beneficios en la investigación biomédica que generaría tal proyecto, fue publicado en la revista Science en 1986 por el virólogo Renato Dulbecco, quien en 1975 había ganado el Premio Nobel de Fisiología o Medicina por sus descubrimientos sobre la interacción entre los virus tumorigénicos y el material genético de la célula. En este trabajo, titulado “Un cambio de rumbo en la investigación de cáncer: secuenciación del genoma humano”, el virólogo pone de manifiesto el hecho de que a pesar de que el trabajo con modelos celulares y virales, así como la aplicación de metodologías de clonación de DNA, habían permitido la identificación de alteraciones moleculares en las células tumorales, era necesario enfocarse más al análisis del genoma celular con la finalidad de entender de forma más profunda los mecanismos de carcinogénesis. Para lograr lo anterior, Dulbecco planteó dos opciones: continuar tratando de identificar todos los genes asociados al cáncer uno por uno, o secuenciar el genoma completo de una especie modelo la cual, si quisiéramos entender el cáncer en humanos, tendría que ser el genoma humano. Como consecuencia de tal proyecto, planteaba Dulbecco, se podrían identificar y clasificar todos los genes y determinar sus perfiles de expresión en diversos tipos celulares, facilitando la identificación de genes involucrados en la génesis y progresión del cáncer.
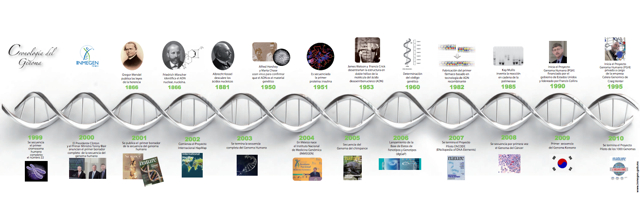
Cronología del genoma. Click en la imagen para ampliar. Fuente: INMEGEN.
Finalmente, el PGH se llevó a cabo gracias a un modelo de colaboración internacional entre seis países, así como al desarrollo de las plataformas de secuenciación automatizada de DNA basadas en el método de Sanger, lo que permitió obtener la secuencia completa del genoma en 13 años y un costo aproximado de $2,700 millones de dólares. Se consideró finalizado en abril del año 2003.
Las predicciones de Dulbecco acerca del impacto de conocer la secuencia del genoma en la investigación y el entendimiento del cáncer han probado ser muy certeras. La disponibilidad de la secuencia del genoma permitió el desarrollo de nuevas plataformas de análisis genómico global, tales como los microarreglos, con los cuales es posible determinar la tasa de expresión de miles de genes en un solo experimento o analizar el número de copias de DNA presente en una muestra de tumor. La aplicación de estas tecnologías al análisis del cáncer permitió estudiar la correlación entre “firmas” moleculares (ya sea a nivel de expresión génica o a nivel de alteraciones en el número de copias de DNA) y variantes clínicas de relevancia, permitiendo el desarrollo de nuevas herramientas de clasificación de los tumores, así como estrategias para evaluar el riesgo de recurrencia de un tumor o su respuesta al tratamiento de forma personalizada para cada paciente con base en las alteraciones genómicas presentes en su tumor.
En el periodo durante el cual se desarrolló el PGH (1990-2003) se continuó con la búsqueda de genes cuya alteración se asociara a diversos tumores humanos. Para el año 2004, un análisis detallado de la literatura llevado a cabo en el Instituto Sanger de Inglaterra (el catálogo COSMIC), identificó 291 genes cuyas mutaciones se asociaban al cáncer. El 90% de los genes de este censo estaban involucrados en translocaciones cromosómicas (intercambio y fusión entre dos cromosomas no relacionados), una situación que se explica dada la relativa facilidad de identificar estas alteraciones mediante técnicas citogenéticas. De esta forma, durante los primeros cinco años del siglo XXI, a pesar de la probada utilidad de las tecnologías existentes, nadie había podido analizar la presencia de mutaciones más que unos cuantos genes en un paciente determinado.
El Proyecto del Genoma del Cáncer Humano
En el año 2004, los institutos Nacionales de Cáncer y de Investigación del Genoma Humano de Estados Unidos llevaron a cabo un taller en el que se exploró la factibilidad de llevar a cabo un proyecto que permitiera obtener un catálogo de la mayor parte de las alteraciones genómicas presentes en tumores humanos. Las conclusiones de esta reunión se presentaron en febrero de 2005, en un documento en el que se describen las recomendaciones para iniciar un “Proyecto del Genoma del Cáncer Humano”. En este documento se detalla el panorama general acerca del conocimiento de las bases genómicas del cáncer y se hace un especial énfasis en los beneficios y aplicaciones clínicas que se han logrado gracias a un mejor entendimiento de las bases moleculares del cáncer y del conocimiento de las alteraciones genómicas presentes en diversos tumores. Entre los ejemplos más exitosos de estas aplicaciones están el desarrollo de fármacos dirigidos para bloquear la acción |
Como el PGH 15 años atrás, el Proyecto del Genoma del Cáncer Humano generó una variedad de críticas, poniendo nuevamente en duda su factibilidad técnica y su utilidad. | |
|
||
Como el PGH 15 años atrás, el Proyecto del Genoma del Cáncer Humano generó una variedad de críticas, poniendo nuevamente en duda su factibilidad técnica y su utilidad. Las críticas se centraban en su tamaño y costo, se decía que el proyecto era comparable a 12 proyectos del genoma humano y que costaría aproximadamente doce mil millones de dólares llevarlo a cabo. Lo que no se tomó en consideración en estas críticas, como sucedió también en el caso del PGH, fue que las tecnologías de secuenciación de DNA sufrirían una transformación radical entre los años 2004 y 2006 gracias al desarrollo de las tecnologías de secuenciación de DNA de nueva generación. Estas nuevas plataformas son capaces de generar la misma información que se obtuvo en todo el Proyecto del Genoma Humano en dos semanas, con un costo aproximado de $10,000 USD, situación que hizo realmente factible poder comparar la secuencia completa de DNA de un tumor y cotejarlo con el genoma normal del mismo paciente para identificar la mayor parte de las mutaciones somáticas presentes en el tejido neoplásico en un tiempo razonable y con un costo relativamente accesible.
Aun antes de la introducción de la secuenciación de nueva generación, varios grupos habían empezado ya a analizar la presencia de mutaciones de forma sistemática en un número limitado de tumores. Usando amplicones generados por reacción en cadena de la polimerasa y secuenciación en capilares, el grupo de Bert Vogelstein publicó en el 2007 un reporte en el que se secuenciaron los exones de los 18,000 genes anotados a la fecha en el genoma humano en 11 tumores de mama y en 11 de colon y recto. Este abordaje permitió obtener por primera vez un panorama completo acerca de las mutaciones presentes en la totalidad de la secuencia codificante de un tumor, lo que eliminó el sesgo introducido por la búsqueda dirigida de mutaciones conocidas y permitió analizar las mutaciones en el contexto de vías celulares completas.
La genómica del cáncer
- Los diferentes tipos de cáncer son un grupo de enfermedades que tienen características similares. Estudiar su origen y analizar su estructura genómica con la finalidad de determinar las alteraciones comunes servirá para encontrar nuevas formas de diagnótico, pronóstico y tratamiento del mismo.
- Fuente: INMEGEN.
En el año 2008 se publicó en la revista Nature el primer reporte de la comparación a nivel de genoma completo entre un tumor y su contraparte normal. Se trató de un caso de leucemia mieloide aguda con un cariotipo normal, la cual se comparó contra el genoma proveniente de células de la piel del mismo individuo. Esta prueba permitió identificar, de principio, que el tumor había adquirido mutaciones en 10 genes, las cuales no estaban en el DNA normal. Dos de estas mutaciones estaban ya previamente reportadas como asociadas a la leucemia, mientras que las ocho restantes nunca habían sido reportadas previamente. Este abordaje demostró que la secuenciación completa del genoma de un tumor y de su contraparte normal constituye una herramienta sin sesgos para identificar alteraciones genómicas previamente no conocidas en genes asociados al cáncer, de una forma eficiente y con costos y tiempos razonables. Este trabajo fue seguido rápidamente por otros que analizaban diversos tumores, incluyendo cáncer de pulmón, mama, melanoma, próstata, ovario y otros, por lo general, reportando todavía mutaciones en un número limitado de muestras.
Una vez demostrada la factibilidad metodológica y analítica de los métodos de secuenciación y detección de mutaciones a nivel de genoma completo, el proyecto planteado por los Institutos Nacionales de Salud de Estados Unidos se convierte en la iniciativa del Atlas del Genoma del Cáncer (TCGA por sus siglas en inglés. http://cancergenome.nih.gov/) y el esfuerzo internacional del ICGC se concreta. Estos dos esfuerzos tienen como objetivo el secuenciar un alto número de muestras (al menos 500 de cada tumor), lo cual representa diversos retos, los cuales incluyen: disponibilidad de muestras adecuadas para el estudio, que hayan sido colectadas bajo un consentimiento informado que permita la secuenciación de genoma completo y que cuenten con datos clínicos adecuados, además de que
Más información
En sus reportes mas recientes, publicados en diversas revistas científicas a lo largo del 2013, el TCGA reporta el análisis conjunto de las alteraciones genómicas más comunes en 21 tipos tumorales distintos, obtenido a partir del análisis de aproximadamente 7,000 tumores, todos ellos analizados mediante secuenciación de exoma o genoma completo y comparados contra tejido normal en cada caso. A partir de este análisis se identificaron cerca de 5 millones de mutaciones puntuales presentes en los tumores, las cuales pueden ser clasificadas en al menos 20 “firmas” de mutación, algunas de ellas presentes en la mayoría de los tumores y otras presentes en tipos tumorales específicos. Además del análisis de secuenciación, el TCGA utiliza diferentes plataformas de análisis genómico, que incluyen el análisis de alteraciones en el número de copias de DNA mediante microarreglos de alta densidad, análisis de transcriptoma y definición de perfiles de mutilación, con la finalidad de ofrecer una visión integral de las alteraciones genómicas en los tumores.
En el caso del ICGC, a la fecha se han registrado en el consorcio 71 proyectos de 67 grupos de investigación en cuatro continentes. En conjunto, estos grupos están analizando más de 25,000 genomas de tumores. Después de la participación en la reunión inicial del ICGC en Toronto y gracias a una colaboración entre el Instituto Carlos Slim de la Salud, el Instituto Nacional de Medicina Genómica y el Instituto Broad de Harvard MIT, México aportó al ICGC el análisis de tumores de mama, cabeza y cuello, y linfoma.
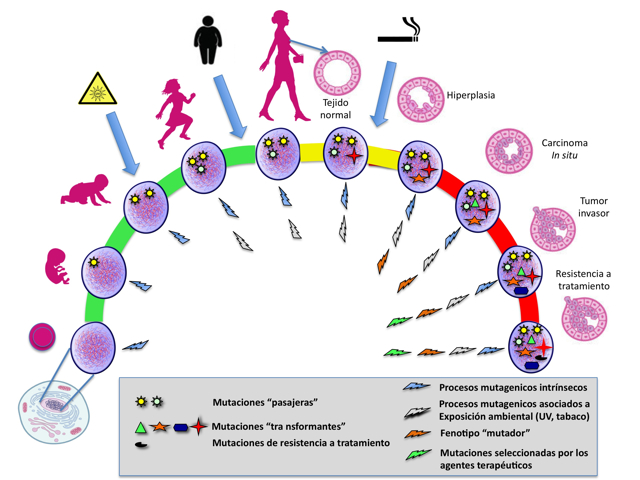
Figura 1. El cáncer surge debido a la acumulación de mutaciones en el genoma a lo largo del tiempo. Desde las primeras divisiones celulares posteriores a la fecundación del óvulo por el espermatozoide, el genoma está expuesto a la acumulación de alteraciones debido a procesos mutacionales intrínsecos, tales como errores en la replicación del DNA. Estos procesos intrínsecos están presentes a lo largo de toda nuestra vida (rayos azules). Después del nacimiento y durante la infancia nos exponemos a otras fuentes potenciales de mutación, tales como la exposición a agentes ambientales como los rayos UV del Sol, lo cual puede ir generando mutaciones adicionales. En la vida adulta, nuestro estilo de vida y aspectos como el tabaquismo y la obesidad elevan considerablemente el riesgo de acumular mutaciones (rayos grises), las cuales pueden desembocar en la formación de un tumor. Las mutaciones en el genoma pueden no resultar en la formación de un tumor si no afectan a genes relacionados con el control del crecimiento y la división celular (mutaciones pasajeras). Sin embargo, si por azar las mutaciones afectan a genes asociados a estos procesos (mutaciones transformantes), se inicia el desarrollo de un tumor, primero como una enfermedad benigna, la cual puede presentar una tasa de mutación más elevada (rayos naranjas), lo que le permitirá acumular mutaciones que resultarán en un tumor invasor.
La caracterización detallada del genoma de los tumores ha tenido un profundo impacto en nuestro entendimiento de las bases moleculares de la carcinogénesis (figura 1). Muchos de estos hallazgos se han traducido en herramientas de aplicación clínica que han mejorado el manejo de los pacientes oncológicos a través del desarrollo de mejores herramientas para la clasificación de los tumores, las cuales también han permitido implementar pruebas de evaluación de riesgo personalizado de recaída o de respuesta a tratamientos particulares.
En resumen, desde las observaciones de Von Hansemann hasta los esfuerzos de secuenciación de genoma completo en cáncer, queda claro que una de las áreas de la biomedicina que más se ha beneficiado de la revolución genómica ha sido la del estudio del cáncer. Sin el desarrollo de las herramientas de análisis genómico, no hubiera sido posible identificar las bases moleculares de muchas de las características que ahora asociamos a la célula neoplásica. El reto ahora está en traducir de forma eficiente y rápida los hallazgos derivados del análisis genómico a aplicaciones de uso clínico y terapéutico.
Aún quedan muchas preguntas acerca de la funcionalidad de muchas de las alteraciones genéticas que se han descrito en los tumores humanos. Sin embargo, una cosa queda clara: durante los próximos cinco a diez años el manejo de los pacientes con cáncer estará fundamentado en gran medida sobre la base de un análisis detallado de las alteraciones genómicas de los tumores, lo que permitirá diseñar tratamientos dirigidos, menos tóxicos y mucho más eficaces para el beneficio de los pacientes con cáncer.
COMENTARIOS
SÍGUENOS









Sitios de interés
ARTÍCULOS RELACIONADOS


UNIVERSIDAD NACIONAL AUTÓNOMA DE MÉXICO
Dirección General de Cómputo y de Tecnologías de Información y Comunicación
Dirección General de Cómputo y de Tecnologías de Información y Comunicación
2013 Esta obra está bajo una licencia de Creative Commons 
