
Desarrollo de nuevos fármacos por computadora
Vol. 22, núm. 6 noviembre-diciembre 2021
Desarrollo de nuevos fármacos por computadora
Martiniano Bello CitaResumen
En este artículo se aborda la utilidad de los métodos computacionales para la identificación y diseño de nuevos compuestos para el tratamiento de enfermedades. Asimismo, se describen los pasos cruciales para el empleo de estos métodos y se mencionan ejemplos de diferentes medicamentos de uso comercial, que han sido diseñados mediante dichas estrategias.
Palabras clave: diseño de fármacos, acoplamiento molecular, tamizaje virtual, dinámica molecular.
Computer development of new drugs
Abstract
This article describes the usefulness of computational methods for the identification and design of new compounds for disease treatment. The crucial steps for these methods are described and examples of different drugs of commercial use, designed using these computational strategies, are mentioned.
Keywords: drug design, molecular docking, virtual screening, molecular dynamics.
Los fármacos y la prevención de enfermedades
A lo largo de los años, se han desarrollado múltiples fármacos que nos han permitido tratar y prevenir enfermedades, con cierta eficacia y seguridad. Comúnmente, el diseño de nuevas sustancias está directamente relacionado con la identificación de compuestos químicos de origen natural, que posteriormente se modifican mediante métodos experimentales para mejorar sus propiedades químicas y poder ser empleados como fármacos. Aunque esta estrategia aún está en uso, el desarrollo de fármacos empleando sólo métodos experimentales involucra una serie de fases que toman alrededor de 15 años y requiere un gasto de entre 500 a 1000 millones de dólares.
En cambio, el diseño de nuevos fármacos por computadora inicia al examinar la información experimental disponible y, a partir de ella, generar modelos predictivos de nuevos compuestos con potencial actividad. Después, éstos tendrán que validar su capacidad como fármacos mediante estudios experimentales. Otra estrategia es el reposicionamiento de fármacos, que es el uso terapéutico de fármacos disponibles en el mercado para tratar una enfermedad distinta. Este método ha mostrado agilizar la aprobación de un nuevo tratamiento, puesto que ya no se requieren estudios de las fases preclínicas y clínicas, que consumen bastante dinero y tiempo (Paul et al., 2013).
En este artículo se presentan los principales métodos y técnicas computacionales auxiliares en el proceso de desarrollo de fármacos por computadora. Además, se discuten ejemplos de proyectos enfocados al diseño de fármacos, desarrollados por un grupo de investigación especializado en la materia.
Aplicación de métodos computacionales en el diseño de fármacos
El diseño de fármacos por computadora se basa en la unión de una serie de disciplinas científicas, que abarcan el modelado molecular, quimioinformática, química teórica y química computacional. Tiene como meta el diseño, optimización y selección de compuestos con actividad biológica. A diferencia del método tradicional, basado en ensayo y error, el método computacional permite reducir el tiempo de obtención de nuevos compuestos, pues es más dirigido y preciso. Por esta razón, es cada vez más común la incorporación de estas herramientas computacionales en la industria farmacéutica y en centros de investigación pública y privada. No obstante, aunque el poder de cómputo junto con los modelos predictivos de nuevos fármacos son notables –al probar su utilidad mediante varios ejemplos de fármacos en el mercado–, aún es necesaria mayor investigación para generar información más precisa.
Cribado virtual y reposicionamiento de fármacos
La primera etapa en el diseño de fármacos o el de identificar fármacos aprobados para una enfermedad que puedan ser empleados para otra distinta (reposicionamiento de fármacos) (Ashburn et al., 2004) consta de manera general de dos pasos: preparación de las estructuras o curado y el cribado virtual.
El cribado virtual o filtrado computacional consiste en construir la base de datos o quimioteca de las estructuras químicas de interés o de la selección de ligandos disponibles comercialmente, para realizar estudios de reposicionamiento de fármacos (ver figura 1). Aquí se seleccionan los compuestos con las mejores propiedades farmacológicas y de afinidad hacia la diana terapéutica (proteína o ácido nucleico) (Lavecchia et al., 2013). Las propiedades farmacológicas se evalúan mediante programas computacionales que nos permiten calcular la absorción, distribución, metabolismo, excreción y perfil toxicológico (predicciones admet1) de un compuesto. Esto se traduce en la inferencia de que ciertos compuestos tengan el potencial de llegar a ser un fármaco.
La afinidad se evalúa inicialmente mediante estudios de acoplamiento molecular o docking (ver figura 1). El docking consiste en predecir la afinidad entre un compuesto y la diana farmacológica (Bello et al., 2013). Estos estudios sólo se realizan a los compuestos que mostraron buenos resultados en las predicciones admet. Finalmente, los compuestos con los mejores valores de afinidad se seleccionan para realizarles estudios de dinámica molecular, que consisten en reevaluar las interacciones para los mejores complejos, entre los compuestos y la diana terapéutica, en un ambiente experimental más parecido al presente en una célula animal. Esto nos permite observar si las interacciones predichas por docking se mantienen, mejoran o desaparecen. Después, los complejos cuyas interacciones se conservan o mejoran son sometidos a un último análisis, que permite reevaluar la afinidad del compuesto por la diana farmacológica. Este cribado virtual puede repetirse varias veces en retroalimentación con los ensayos experimentales, para refinar los resultados.
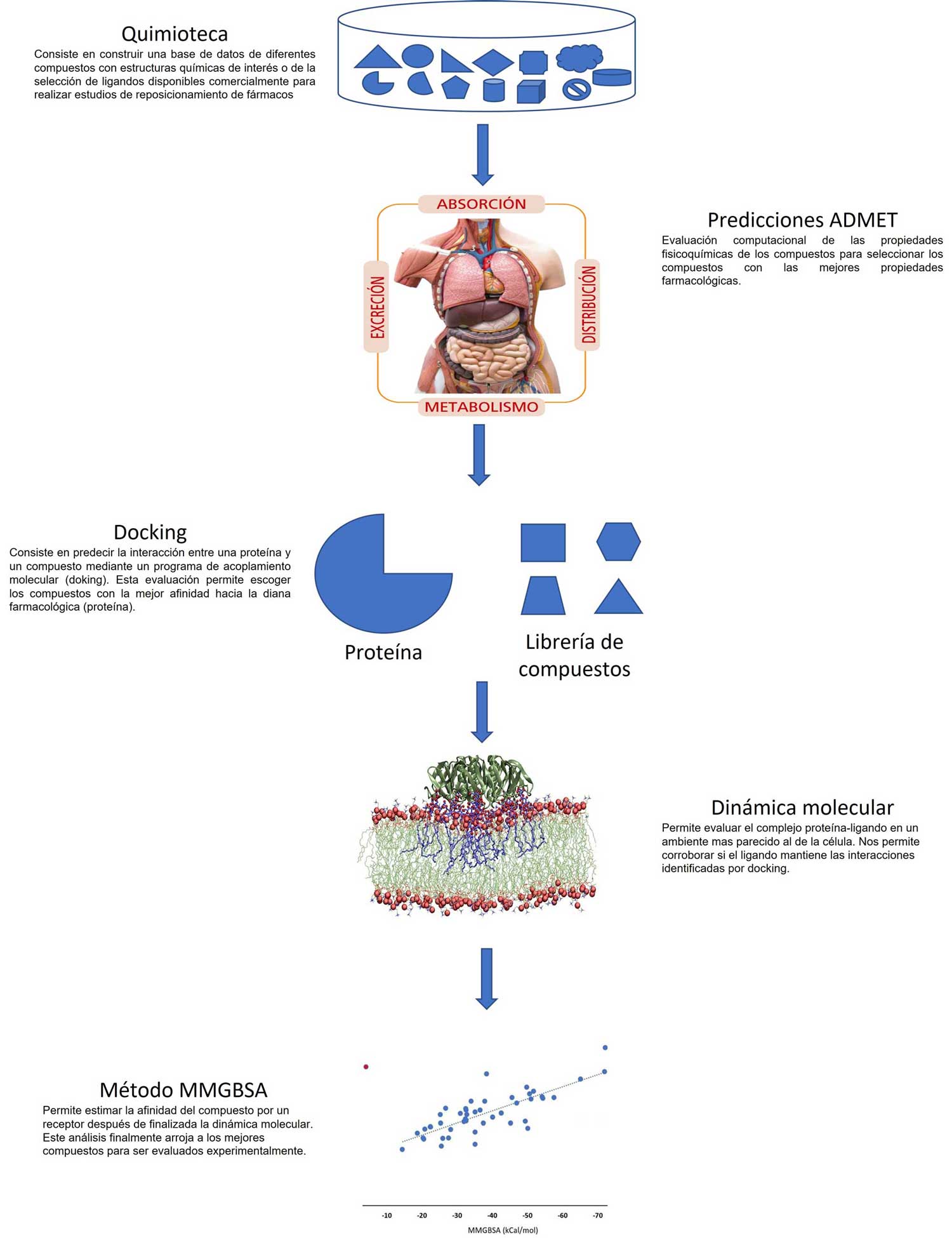
Figura 1. Serie de pasos que se siguen en un grupo de investigación enfocado en el diseño de fármacos por computadora.
Docking y dinámica molecular
Aunque un fármaco potencial debe cumplir con buenas propiedades admet, lo más importante es que muestre una alta afinidad y selectividad hacia la diana terapéutica, que es una molécula clave involucrada en una vía metabólica particular y que está asociada con una condición patológica. En este contexto, el predecir la afinidad entre una proteína y un compuesto mediante estudios de docking ha sido ampliamente utilizado para el cribado virtual a gran escala, en una quimioteca de miles de compuestos dirigidos hacia dianas terapéuticas específicas (Murcko et al., 1995).
Actualmente, el docking tiene un éxito limitado. No obstante, a partir de éste, se han empleado otros métodos computacionales más robustos, que permiten estimar la afinidad entre un ligando y una diana farmacológica. La dinámica molecular tiene el potencial de permitirnos reevaluar las interacciones predichas por docking para un complejo proteína-ligando, en un ambiente biológico más parecido al de la célula. La dinámica molecular combinada con cálculos de energía de asociación, como el método de Mecánica Molecular-Generalizada Born/Área de Superficie (mmgbsa) (Miller et al., 2012), permite estimar la afinidad del compuesto por un receptor, después de finalizada la dinámica molecular. Al final, este análisis identifica los mejores compuestos para ser evaluados experimentalmente.
Ejemplos
Existen varios casos exitosos del diseño de nuevos fármacos por computadora. Por ejemplo, la dorzolamida se emplea en el tratamiento de una enfermedad ocular que daña el nervio óptico y es el primer fármaco diseñado por computadora, en 1995. Otros ejemplos son el imatinib, cimetidina, raltegravir y zanamivir (ver figura 2). De ellos, el imatinib es usado para tratar la leucemia mieloide crónica y otros tipos de cáncer; la cimetidina es empleada para tratar úlceras gastroesofágicas; el raltegravir es un antiviral para tratar los síntomas del virus de la inmunodeficiencia humana (vih), mientras que zanamivir es utilizado en el tratamiento de la gripe común.
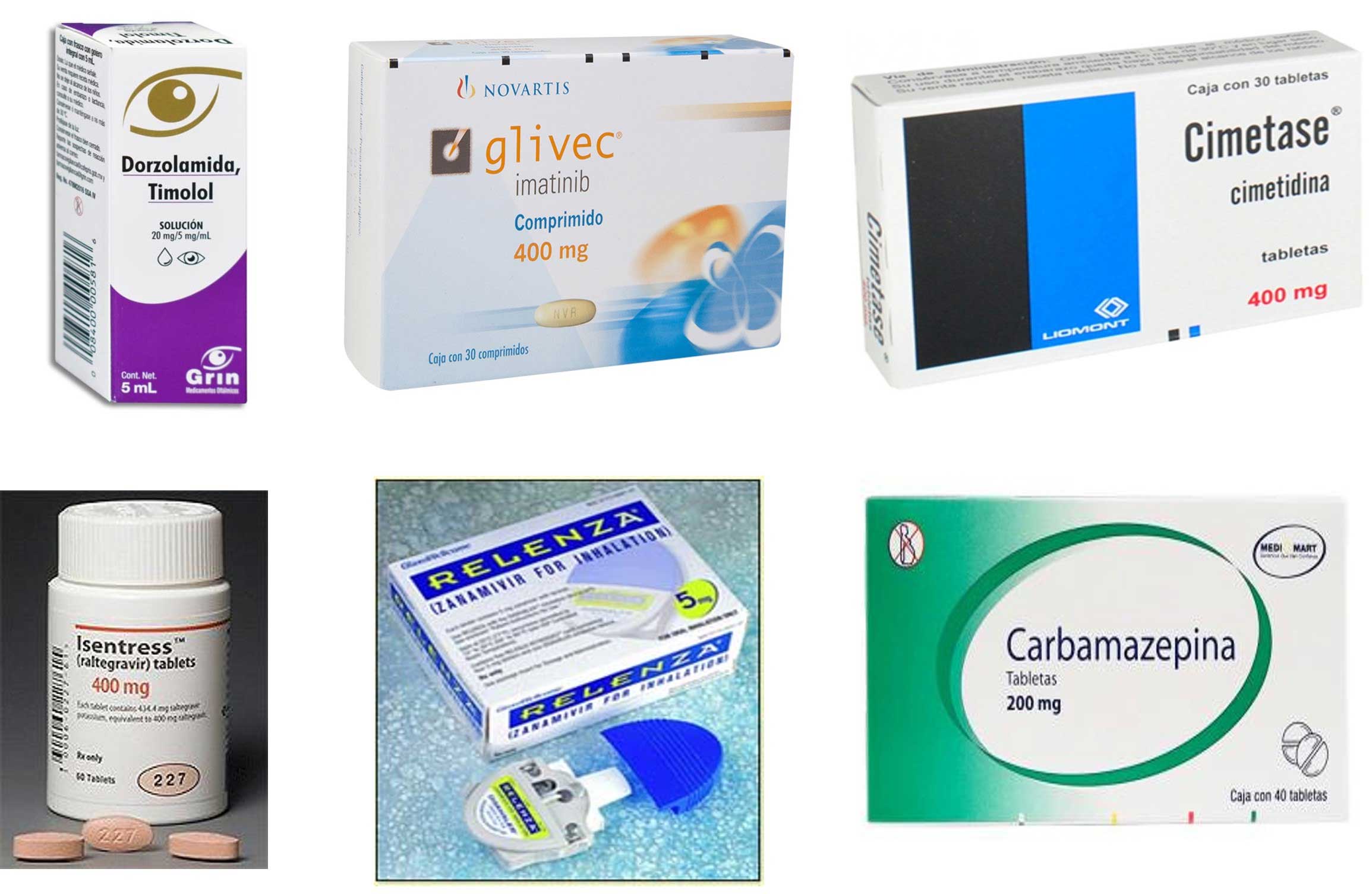
Figura 2. Diferentes medicamentos que contienen a los fármacos dorzolamida, imatinib, cimetidina, raltegravir, zanamivir, carbamazepina.
Un ejemplo de reposicionamiento de fármacos en México fue desarrollado por investigadores del Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán. Ellos identificaron a la carbamazepina (ver figura 2), que es empleada para tratar la epilepsia, y como un fármaco loable para tratar problemas asociados a la diabetes (Rull et al., 1969).
Investigación en el diseño de fármacos
Hay varios grupos de investigación en México y el mundo especializados en el desarrollo y aplicación de metodologías para el diseño de fármacos por computadora. En México, en mi grupo de trabajo, se realiza investigación en el desarrollo de modelos teóricos computacionales, así como la búsqueda de nuevos compuestos útiles para inhibir macromoléculas que participan en el crecimiento y funcionamiento celular. Cuando estas macromoléculas funcionan de manera incorrecta, se presentan graves consecuencias en la salud, por ejemplo, el desarrollo de diferentes tipos de cáncer.
A la fecha, y en colaboración con otros investigadores, hemos logrado identificar diferentes ligandos con potencial uso contra el cáncer de páncreas. Adicionalmente, estudios de reposicionamiento de fármacos nos han permitido identificar diferentes compuestos comerciales con potencial uso contra el cáncer de mama. Así, con el objetivo de desarrollar nuevos fármacos, hemos establecido cuatro estrategias computacionales en nuestro grupo de investigación:
- Entender las relaciones estructura-actividad biológica de compuestos inhibidores reportados.
- Identificar nuevos inhibidores naturales o sintéticos.
- Optimizar compuestos líderes.
- Reposicionamiento de fármacos aprobados por la Administración de Medicamentos y Alimentos de Estados Unidos (Food and Drug Administration, fda).
La primera estrategia consiste en recabar información del sistema a estudiar, mediante estudios de modelado molecular de inhibidores ya conocidos. Los métodos que se ocupan en esta fase son el acoplamiento molecular, dinámica molecular combinada con el método mmgbsa. La segunda estrategia es el cribado virtual de bases de datos moleculares para la identificación de nuevos inhibidores. El cribado virtual de librerías de compuestos naturales y sintéticos, emplea principalmente acoplamiento molecular para seleccionar los compuestos más promisorios, y posteriormente dinámica molecular y mmgbsa para refinar los resultados de afinidad. Este cribado ha permitido tener bibliotecas de compuestos que pueden ser dirigidos hacia otras dianas terapéuticas. La tercera estrategia es el diseño de nuevos inhibidores, fundado en los complejos ligando-proteína recabados por la segunda estrategia. Su objetivo es proponer modificaciones químicas en los nuevos inhibidores, que incrementen las interacciones con su receptor. La cuarta estrategia es el reposicionamiento, para identificar fármacos aprobados para una enfermedad que puedan ser empleados para otra distinta.
Desarrollo de inhibidores de histonas deacetilasas
En el cáncer de tejidos que conforman la sangre (leucemia) existe una alta concentración de diferentes proteínas denominadas histonas deacetilasas (hdacs).2 Esto favorece que se inactiven de manera preferente los genes que ayudan a detener el desarrollo del cáncer, favoreciendo el desarrollo de esta enfermedad. En particular, se ha identificado que diferentes histonas se encuentran en altas concentraciones en distintos tipos de leucemias. Así, se plantea que el diseño de inhibidores de hdacs (ihdacs) es una estrategia para incrementar la activación de genes que ayuden a revertir el cáncer (Robey et al., 2011).
Hoy en día se encuentran en uso clínico cuatro fármacos, que comparten estructuras químicas similares, aprobados por la fda para el tratamiento de diferentes tipos leucemias. Sin embargo, su empleo está ligado a efectos colaterales graves como problemas gastrointestinales y de coagulación. Es por ello por lo que hay un especial interés para identificar y desarrollar nuevos ihdacs que tengan una estructura química más selectiva hacia ciertas hdacs y con efectos secundarios menos graves. A la fecha se han encontrado varios ihdacs por diversos métodos y con orígenes diferentes, incluyendo productos naturales (Seidel et al., 2012), fármacos obtenidos por reposicionamiento (Moreira-Silva et al., 2020), y compuestos de origen sintético (Sixto-López et al., 2020).
Dentro de las técnicas más utilizadas para la identificación de nuevos ihdacs se encuentran el acoplamiento molecular (Scafuri et al., 2020) y la dinámica molecular (Sixto-López et al., 2020). En nuestro grupo, se han diseñado una serie de compuestos con estructura química similar a los ihdacs, aprobados para uso clínico (Sixto-López et al., 2020). Para estas investigaciones, inicialmente se recabó información de complejos biológicos entre ihdacs y tres diferentes hdacs, mediante estudios de acoplamiento y dinámica molecular. En la segunda fase se diseñó una librería de nuevos ihdacs, basada en la información de la etapa inicial. De esta biblioteca, se seleccionaron los compuestos con las mejores propiedades de unión a las tres hdacs estudiadas, a través de estudios de acoplamiento y dinámica molecular. Finalmente, se evalúo la actividad biológica de los compuestos con estudios en líneas celulares de cáncer. Basándonos en éstos, se demostró el potencial anticáncer de los compuestos con las mejores propiedades de unión.
Reposicionamiento de inhibidores del receptor del factor de crecimiento epidérmico
El receptor 1 del factor de crecimiento epidérmico humano (epithelial growth factor receptor, egfr, por sus siglas en inglés) y el receptor 2 del factor de crecimiento epidérmico humano (epithelial growth factor receptor 2, her2, por sus siglas en inglés) participan en diversos mecanismos en la célula, que controlan su multiplicación y supervivencia. En 40% de los casos de cáncer de mama existe una alta concentración de her2 y ausencia de receptores estrogénicos, lo que hace que las células se multipliquen más rápido de lo normal. Este tipo de enfermedad es denominada cáncer her2+ y hoy en día se encuentran en uso clínico tres fármacos contra ella: neratinib, lapatinib y tucatinib (ver figura 3). No obstante, su empleo está asociado a efectos secundarios importantes, así como a un alto costo.

Figura 3. Diferentes medicamentos que contienen a los fármacos tucatinib, neratinib y lapatinib.
Por lo anterior, la búsqueda de nuevos inhibidores de her2, con efectos colaterales menos drásticos está justificada. Actualmente se han identificado diferentes inhibidores de origen natural (Wang et al., 2016), obtenidos por reposicionamiento (Balbuena-Rebolledo et al., 2021), y otros de origen sintético (Bello et al., 2020), empleando cribado virtual, acoplamiento y dinámica molecular.
Un ejemplo de un estudio, que se realizó con la colaboración de nuestro grupo de investigación, consistió en demostrar que tres fármacos aprobados por la fda para tratar otras enfermedades también pueden actuar como inhibidores duales de her2 y egfr (Balbuena-Rebolledo et al., 2021). Primero, se realizaron análisis para entender las bases energéticas y estructurales de complejos biológicos entre inhibidores conocidos de her2 y egfr, a través de estudios de acoplamiento y dinámica molecular. En la segunda fase se hizo una búsqueda en una base de datos de compuestos aprobados por la fda, empleando el criterio de similaridad estructural con lapatinib. De la lista de compuestos seleccionados se procedió a realizar estudios de acoplamiento y dinámica molecular, para seleccionar los compuestos más prometedores. Posteriormente, se evalúo su actividad biológica en líneas celulares de cáncer de mama. Finalmente, se demostró el potencial anticáncer de los compuestos reposicionados.
Conclusiones
A pesar de la falta de precisión que aún se mantiene en los métodos computacionales para describir sistemas complejos, éstos han ganado terreno debido a su utilidad y versatilidad. Aunque su empleo puede ser entendido como “sencillo”, y que se podría llevar a cabo sin conocimiento químico, biológico e informático suficiente del sistema a estudiar, es importante recalcar que, para un apropiado desarrollo de un proyecto empleando las técnicas computacionales descritas en este artículo, debe existir un balance adecuado entre el conocimiento científico y el impacto farmacológico, con la finalidad de poder decidir de manera racional la relevancia de la información y poder proceder hacia la fase experimental. Finalmente, las predicciones obtenidas mediante métodos computaciones necesitan ser puestas a prueba experimentalmente, para validar los hallazgos computacionales.
Referencias
- Ashburn, T. T. y Thor, K. B. (2004). Drug repositioning: Identifying and developing new uses for existing drugs. Nature Reviews Drug Discovery, 3, 673-683. https://doi.org/10.1038/nrd1468.
- Balbuena-Rebolledo, I., Padilla-Martínez, I. I., Rosales-Hernández, M. C. y Bello, M. (2021). Repurposing fda Drug Compounds against Breast Cancer by Targeting egfr/her2. Pharmaceuticals, 14(8), 791. https://doi.org/10.3390/ph14080791.
- Bello, M., Martinez-Archundia, M. y Correa-Basurto, J. (2013). Automated docking for novel drug discovery. Expert Opinion on Drug Discovery, 8(7), 821-834. https://doi.org/10.1517/17460441.2013.794780.
- Bello, M., Guadarrama-García, C. y Rodriguez-Fonseca, R.A. (2020). Dissecting the molecular recognition of dual lapatinib derivatives for egfr/her2. J Comput Aided Mol Des, 34(3), 293-303. https://doi.org/10.1007/s10822-019-00270-4.
- Miller iii, B. R., McGee Jr, T. D., Swails, J. M., Homeyer, N., Gohlke, H. y Roitberg, A. E. (2012). mmpbsa.py: an efficient program for end-state free energy calculations. Journal of chemical theory and computation, 8(9), 3314-3321. https://doi.org/10.1021/ct300418h.
- Lavecchia, A. y di Giovanni, C. (2013). Virtual screening strategies in drug discovery: A critical review. Current Medicinal Chemistry, 20(23), 2839-2860. https://doi.org/10.2174/09298673113209990001.
- Li, J., Wang, H., Li, J., Bao, J. y Wu, C. (2016). Discovery of a Potential her2 Inhibitor from Natural Products for the Treatment of her2-Positive Breast Cancer. Int J Mol Sci, 17(7), 1055. https://doi.org/10.3390/ijms17071055.
- Moreira-Silva, F., Camilo, V., Gaspar, V., Mano, J.F., Henrique, R., Jerónimo, C. (2020). Repurposing Old Drugs into New Epigenetic Inhibitors: Promising Candidates for Cancer Treatment? Pharmaceutics, 12(5), 410. https://doi.org/10.3390/pharmaceutics12050410.
- Murcko, M. A. (1995). Computational methods to predict binding free energy in ligand-receptor complexes. Journal of medicinal chemistry, 38(26), 4953-4967. https://doi.org/10.1021/jm00026a001.
- Paul, S. M. y Lewis-Hall, F. (2013). Drugs in search of diseases. Science Translational Medicine, 5(186), 186fs18. https://doi.org/10.1126/scitranslmed.3004452.
- Rull, J. A., Quibrera, R., González-Millán, H. y Castañeda Lozano, O. (1969). Symptomatic treatment of peripheral diabetic neuropathy with carbamazepine (Tegretol®): Double blind crossover trial. Diabetologia, 5, 215-218.
- Seidel, C, Schnekenburger, M., Dicato, M. y Diederich, M. (2012). Histone deacetylase modulators provided by Mother Nature. Genes Nutr, 7(3), 357-367. https://doi.org/10.1007/s12263-012-0283-9.
- Scafuri, B., Bontempo, P., Altucci, L., De Masi, L. y Facchiano, A. (2020). Molecular Docking Simulations on Histone Deacetylases (hdac)-1 and -2 to Investigate the Flavone Binding. Biomedicines, 8(12), 568. https://doi.org/10.3390/biomedicines8120568.
- Sixto-López, Y., Gómez-Vidal, J. A., de Pedro, N., Bello, M., Rosales-Hernández, M. C. y Correa-Basurto, J. (2020). Hydroxamic acid derivatives as hdac1, hdac6 and hdac8 inhibitors with antiproliferative activity in cancer cell lines. Sci Rep, 10(10462). https://doi.org/10.1038/s41598-020-67112-4.
Recepción: 24/02/2021. Aprobación: 07/09/2021.