
¿Es posible identificar nuevos fármacos desde tu computadora?
Vol. 25, núm. 1 enero-febrero 2024
¿Es posible identificar nuevos fármacos desde tu computadora?
María Teresa Alvarado Parra, María Gabriela Mancilla Montelongo y Karina Verdel Aranda CitaResumen
¿Cómo se obtienen los nuevos fármacos? ¿De dónde salen nuevos productos para descontaminar un lago o el suelo? Las plantas y los microorganismos producen millones de moléculas que pueden tener una de estas importantes aplicaciones biotecnológicas. Hasta hace unos años, la caracterización biotecnológica era un proceso costoso y complicado. Sin embargo, la revolución de la bioinformática trajo consigo las bases de datos biológicas que, en conjunto con el desarrollo de la tecnología, han permitido el diseño y desarrollo de algoritmos para conocer la función biológica de un compuesto. Gracias a ello, actualmente se puede predecir la actividad biológica de una gran variedad de moléculas antes de emprender un experimento en el laboratorio. El objetivo de este artículo es compartir cómo es que se pueden caracterizar productos naturales, como fármacos, desde una computadora personal, utilizando bases de datos pertinentes y métodos computacionales in silico, para después comprobar la actividad biológica con pruebas in vitro.
Palabras clave: base de datos, in silico, acoplamiento molecular, productos naturales, bioinformática.
Is it possible to identify new drugs from your computer?
Abstract
How are new drugs obtained? Where do new products to decontaminate a lake or soil come from? Plants and microorganisms produce millions of molecules that can have one of these important biotechnological applications. Until a few years ago, biotechnological characterization was an expensive and complicated process. However, the bioinformatics revolution brought with it biological databases that, in conjunction with the development of technology, have allowed the design and development of algorithms to know the biological function of a compound. Thanks to this, it is now possible to predict biological activities from a wide variety of molecules, before undertaking an experiment in the laboratory. The objective of this article is to share that it is possible to characterize natural products, such as drugs, from a personal computer, using relevant databases and in silico computational methods, and then verify the biological activity with in vitro tests.
Keywords: databases, in silico, molecular docking, natural products, bioinformatics.
Introducción
—Hace un año, trabajé en un proyecto para la biorremediación1 del río Atoyac, en Puebla, México. Entre los contaminantes de este río, están los tintes usados en la industria textil, que son altamente tóxicos y poco degradables. Para llevar a cabo dicha biorremediación, los investigadores querían utilizar bacterias que produjeran enzimas capaces de reducir estos compuestos tóxicos. El problema era que no estaban seguros si las enzimas producidas por las bacterias eran capaces de reaccionar de manera específica con esas moléculas. Entonces, mi trabajo fue realizar computacionalmente un acoplamiento molecular de las enzimas bacterianas2 propuestas con cada una de las moléculas tóxicas seleccionadas. Así, si la afinidad de unión3 entre la enzima y el compuesto era buena, quería decir que la enzima era capaz de reducir el compuesto (ver video 1). Una vez realizada la propuesta, el grupo de investigación recopiló los análisis y diseñó un sistema bacteriano capaz de degradar esos compuestos (Acevedo et al., 2021).
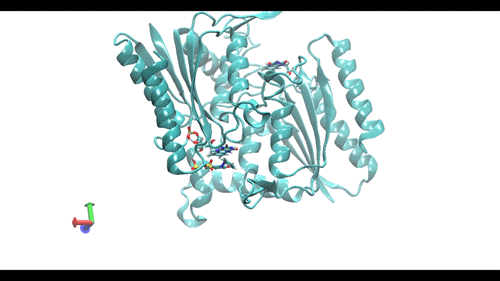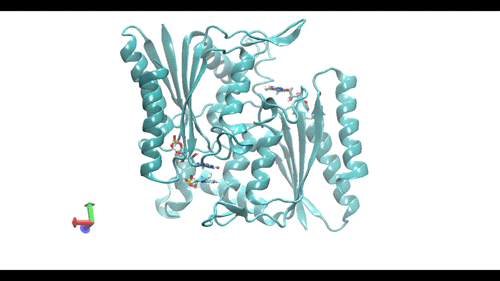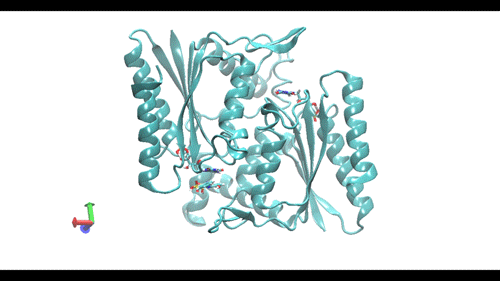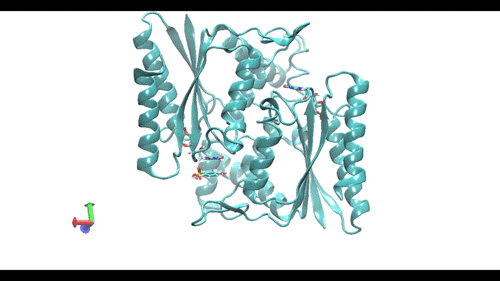
Video 1. Resultado de una simulación dinámica realizada en GROMACS a 10 ns de Pseudomonas putida y el compuesto azoico azul índigo.
Crédito: el resultado se elaboró y visualizó con el software Visual Molecular Dynamics (VMD).
El anterior testimonio, perteneciente a una estudiante de la Licenciatura en Biología, es la punta del iceberg sobre lo que se puede hacer o predecir desde una computadora. En los últimos años, la demanda por los métodos computacionales en numerosas investigaciones ha ido creciendo y, con ello, la importancia de hacer más accesible la bioinformática y la biología computacional en el descubrimiento de nuevos compuestos activos. Por ello, en este artículo se describen métodos computacionales generales para la caracterización biotecnológica. Esto incluye el concepto de acoplamiento molecular de enzimas y ligandos, junto con los pasos que se siguen para llevar a cabo este análisis computacional.
Fuentes de nuevos compuestos y cómo analizar grandes volúmenes de datos
Los productos naturales (pn), en un sentido amplio, se refieren a cualquier parte, biomolécula (como las enzimas o proteínas) o compuesto químico (también llamado metabolito secundario) obtenidos de un ser vivo, como una planta o un microorganismo (Sarker y Nahar, 2012). Una de las aplicaciones más importantes de los pn es en biotecnología ambiental, específicamente en la biorremediación. Las plantas y microorganismos tienen un papel fundamental en el reciclaje de carbono, nitrógeno, fósforo y azufre. Al mismo tiempo, sus pn son capaces de degradar compuestos contaminantes más complejos como los provenientes de las actividades industriales. Las principales aplicaciones de estos pn son el tratamiento de aguas residuales, degradación de xenobióticos4 y petroquímicos (Glazer y Nikaido, 2007). Este tipo de moléculas pequeñas, como los contaminantes o los metabolitos secundarios, se convierten en ligandos cuando interaccionan con las biomoléculas, como las proteínas.
Al recopilar una vasta información taxonómica, molecular, genética, metabólica y toxicológica de pn se ha generado la necesidad de almacenar y poner a disposición grandes volúmenes de datos. El surgimiento de las bases de datos (bd) permitió resolver este problema de almacenamiento y mejoró la disponibilidad de datos biológicos. Hoy en día, la bioinformática es esencial para manejar y comprender esta información. El uso de diferentes métodos que combinan algoritmos, lenguajes de programación, programas y otras herramientas han dado lugar a lo que se conoce como análisis in silico (Cañedo-Andalia y Arencibia Jorge, 2004). Esto quiere decir que se analizan en la computadora las interacciones entre diferentes pn. Con la información que nos proveen las bd es posible predecir la actividad de un pn en tres etapas básicas (ver figura 1).
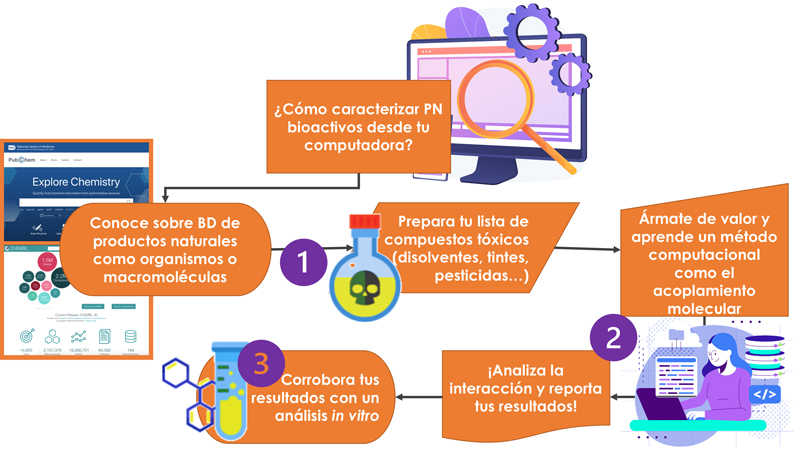
Figura 1. Cómo caracterizar productos naturales activos desde tu computadora, en tres sencillos pasos: 1) conocer las bases de datos, 2) aprender y analizar mediante un método computacional, y 3) corroborar el resultado in silico con un experimento in vitro.
Crédito: imágenes y vectores de Freepik.com.
Paso 1. Bases de datos de productos naturales y de actividad biológica
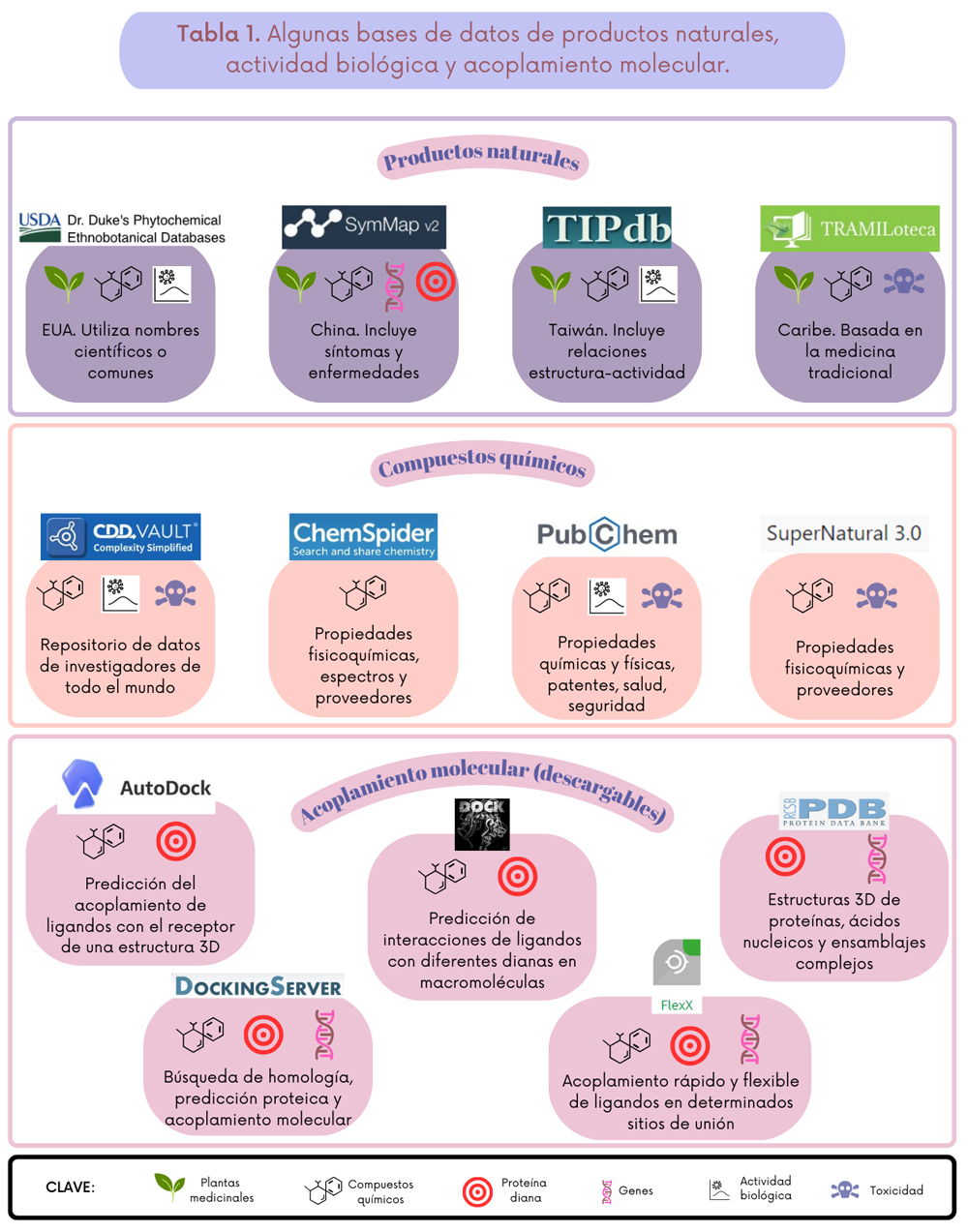
Las bd de productos naturales cambian de acuerdo con la zona geográfica, tipo de organismo (hierbas, plantas vasculares, cactáceas, etcétera), propiedad biológica, uso y aplicación (antibióticos, anticancerígenos, suplementos alimenticios, entre otros), compuestos químicos o proteínas. Otro tipo de bd son como bibliotecas virtuales que resguardan artículos especializados y permiten acceder a información relevante para elegir un modelo de estudio. Las bd enfocadas en actividad biológica almacenan propiedades de interacción entre moléculas (compuestos químicos-proteínas-genes). La actividad biológica es el potencial que tiene una molécula para llevar a cabo una función, por ejemplo, su uso contra una determinada enfermedad (Jackson et al., 2007). Algunas bd reportan datos como concentraciones inhibitorias, dosis efectivas y dosis letales o tóxicas. La mayoría de las bd son de acceso libre (algunos ejemplos se encuentran en el cuadro 1), otras son exclusivas para investigadores y requieren de un registro. También existen bd con un costo, las cuales, en su mayoría son generadas para la industria farmacéutica.
Paso 2. Métodos computacionales: el acoplamiento molecular
El acoplamiento molecular es una predicción de las interacciones entre dos pn: un ligando (molécula pequeña) y una proteína. Los primeros acoplamientos moleculares se basaron en métodos qsar (relaciones cuantitativas estructura–actividad, por sus siglas en inglés) (Ekins et al., 2007). Estos métodos utilizaban computadoras con procesadores hechos de silicio para estimar la actividad biológica de las moléculas; por ello a estos análisis se les llamó in silico. Las primeras predicciones con computadoras usaban modelos matemáticos que relacionaban una estructura con una propiedad fisicoquímica o una actividad biológica, siguiendo como principio la estadística. El primer registro de un fármaco con un diseño in silico es el mesilato de nelfinavir, usado para inhibir una proteína del Virus de Inmunodeficiencia Humana (vih) (Meza Menchaca et al., 2020).
Después surgieron metodologías de acoplamiento basadas en descriptores, reglas, conocimiento, ligandos, objetivos, afinidad virtual, entre muchas otras (Ekins et al., 2007). Dentro de las más usadas se encuentran aquellas que dependen de la disponibilidad de información estructural. Por esta razón, durante un tiempo, los científicos se concentraron en experimentos con las estructuras 3D de proteínas. Todas estas metodologías condujeron a la creación de múltiples interfaces y softwares de afinidad virtual (ver video 1).
Gracias a las numerosas metodologías, se han descubierto inhibidores y dianas terapéuticas que han servido para probar diferentes ligandos que prometen ser futuros fármacos. Es fascinante el control en tiempo récord de brotes de enfermedades a partir de la identificación de compuestos bioactivos y su afinidad a las dianas terapéuticas (Meza Menchaca et al., 2020). Un ejemplo bastante familiar fue la búsqueda de fármacos para el tratamiento de covid-19, donde el acoplamiento molecular fue “el rey de los métodos computacionales” (Pavan y Moro, 2023).
El acoplamiento molecular ha evolucionado de manera que nos ha permitido reportar y comprobar interacciones de distinta naturaleza, como interacciones ligando-proteína (ver figura 2 y video 1), proteína-proteína, adn-ligando, adn-proteína, sólido-proteína, sólido-ligando, entre otras (Dar y Mir, 2017).
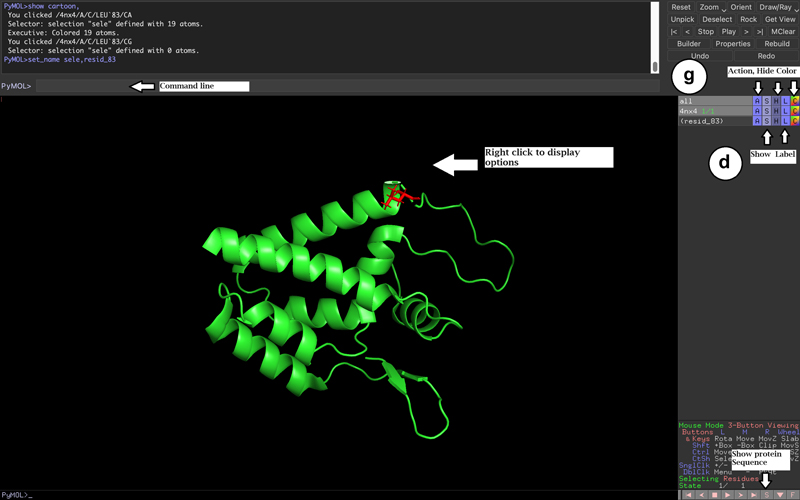
Figura 2. Acoplamiento molecular proteína-ligando. La estructura terciaria de la proteína está marcada en verde y la molécula pequeña (ligando) en rojo. Visualización con el programa PyMol donde, utilizando los botones de los apartados marcados, se puede eliminar las moléculas de agua (paso (d)) y observar las interacciones del acoplamiento (paso (g)).
¿Cómo se puede llevar a cabo un análisis de acoplamiento molecular?
La metodología para realizar los acoplamientos moleculares puede resumirse en siete pasos (ver listado a – g), lo que requiere diferentes bd y programas computacionales (ver figura 1). Cabe destacar que esta secuencia puede considerarse como una receta básica general, pero cada bioinformático la modifica y adapta según sus objetivos y lo que intenta analizar.
- Obtención de estructuras. Consulta de diferentes bd para descargar las estructuras 3D de las moléculas para el acoplamiento (ver figura 1 y tabla 1).
- Búsqueda de dominios funcionales. Una proteína puede tener dominios funcionales5 compartidos con varias familias proteicas, pero esto no significa que mantengan su función biológica. De esta manera, se consultan los dominios funcionales, se anotan y, después del acoplamiento molecular, se verifica si la unión sucedió dentro de estos dominios.
- Exploración de sitios activos. Las proteínas contienen sitios específicos donde otras moléculas pueden unirse y llevar a cabo su función biológica. A estos sitios se les conoce como bolsillos de unión o sitios activos. Éstos se muestran como cavidades en las estructuras 3D de las proteínas donde entrará el ligando.
- Minimización energética de moléculas. A veces las moléculas 3D contienen moléculas de agua u otras con metales pesados. Éstas pueden interferir en el acoplamiento molecular y deben eliminarse. Después, se minimiza la energía de las moléculas 3D para corregir distorsiones o tensiones, reducir el movimiento y garantizar una estructura estable (ver figura 2).
- Acoplamiento molecular. En este paso se seleccionan los aminoácidos que conforman el sitio activo de la proteína y se realiza el acoplamiento molecular de los ligandos (ver figura 2).
- Análisis de resultados. El acoplamiento molecular arrojará varios resultados, pero los más importantes son la energía de enlace (describe la fuerza de interacción proteína-ligando) y el score o puntuación de acoplamiento (refleja la calidad de la unión). Mientras menores sean estos valores, más probable es que la función biológica se cumpla.
- Visualización de interacciones. Se observan las interacciones intermoleculares (fuerzas de van der Waals, interacciones iónicas, etcétera). También se verifica si la unión sucedió dentro de los aminoácidos que conforman los dominios funcionales del paso b (ver figura 2).
¿Por qué se realizan los pasos de minimización y medición de energías en las biomoléculas? Como puede observarse en el video 1, estas biomoléculas no son rígidas y estáticas como rocas. Su interacción con los ligandos para predecir una posible actividad biológica tampoco es como armar legos. Más bien, tienen unas partes que les dan estructura y otras que son como resortes en movimiento. Entonces, se usan ecuaciones matemáticas para predecir cómo se mueven estos resortes, qué carga tienen y cómo interactúan con los ligandos o las moléculas de agua. Justo con las herramientas computacionales se pueden hacer estos cálculos matemáticos y probar muchas condiciones y ligandos de manera eficiente y muy cercana a la realidad.
Paso 3. ¿Análisis in silico o análisis in vitro?
Un experimento in vitro es aquel donde se comprueba si un pn es activo. Se utilizan placas o cajas plásticas o de vidrio (de ahí el término in vitro) y, para que sea válido, se requieren múltiples repeticiones de una misma prueba. Entonces, si ya se realizó el análisis in silico, ¿es necesario realizar un experimento in vitro? ¿Cuándo es necesario hacer un análisis in silico o uno in vitro? Es normal plantearse estas preguntas y la respuesta es más sencilla de lo que parece. Cuando se reporta un nuevo compuesto con determinada actividad para resolver un problema médico o ambiental es común ver un análisis in vitro, en el que el compuesto fue probado a diferentes concentraciones. Sin embargo, los análisis in vitro no revelan la interacción que existe entre los compuestos y, por tanto, no tenemos conocimiento sobre porqué es efectivo y las posibles alternativas. Las investigaciones que usan herramientas in silico permiten entender, en un menor tiempo, la interacción entre compuestos. Por lo tanto, el análisis in silico es un complemento de los análisis in vitro.
El futuro está en las computadoras
En la actualidad, se puede utilizar una computadora personal para dar los primeros pasos en el descubrimiento de compuestos activos a través del acoplamiento molecular. Además, gracias a internet, podemos acceder vía remota a múltiples bd y programas bioinformáticos. Sin embargo, ésta es una visión reducida del potencial que tienen los métodos computacionales. En análisis masivos de datos in silico es ideal la independencia de la web. Para esto se usan computadoras de alta gama que tienen miles de gb de memoria, cientos de tb de almacenamiento y decenas de núcleos de procesamiento. Estos equipos suelen estar conectados entre sí formando clusters,6 que pueden analizar cientos de procesos bioinformáticos al mismo tiempo de manera eficiente.
Conclusión
Aún nos queda mucho por descubrir, pero, gracias a la bioinformática y la biología computacional, los descubrimientos están siendo mucho más rápidos que nunca. En general, los bioinformáticos descubren fármacos desde su computadora en tres pasos: a) conociendo bd sobre organismos y compuestos activos, b) usando métodos computacionales como el acoplamiento molecular y c) combinando métodos in silico e in vitro.
¿Cuáles son las nuevas tendencias y el futuro de esta área de la biología computacional? Debido a la gran cantidad de datos que actualmente existen, conocidos como big data, los investigadores y las compañías farmaceúticas ya están usando inteligencia artificial (ia) para analizar toda esta información molecular. Lo anterior les permite predecir de manera muy precisa nuevas estructuras, lo que ahorra tiempo y dinero. Además, el avance en el desarrollo de métodos computacionales en general se enfoca en abordar los principales retos en esta área, como la alta flexibilidad de las biomoléculas y la lentitud en la unión-disociación de las mismas. Sin embargo, hay temas que aún necesitan respuesta, como el entendimiento de los mecanismos de reacción, los procesos termodinámicos y la cinética de las interacciones moleculares, entre otros.
Referencias
- Acevedo, A., Alvarado, M., Castañon, V., y Diaz, V. (2021). Azoyac: Cleaning Atoyac´s water using synthetic biology. concytep. https://app.jogl.io/es/project/762/Azoyac.
- Cañedo Andalia, R., y Arencibia Jorge, R. (2004). Bioinformática: en busca de los secretos moleculares de la vida. Acimed, 12(6), 1-29. http://tinyurl.com/bde2upvu.
- Dar, A. M., y Mir, S. (2017). Molecular Docking: approaches, types, applications and basic challenges. Journal of Analytical Bioanalytical Techniques, 8, 356. https://doi.org/10.4172/2155-9872.1000356.
- Ekins, S., Mestres, J., y Testa, B. (2007). In silico pharmacology for drug discovery: methods for virtual ligand screening and profiling. British Journal of Pharmacology, 152(1), 9–20. https://doi.org/10.1038/sj.bjp.0707305.
- Glazer, A. N., y Nikaido, H. (2007). Microbial biotechnology: fundamentals of applied microbiology (2a. ed.) Cambridge University Press.
- Jackson, C. M., Esnouf, M. P., Winzor, D. J., y Duewer, D. L. (2007). Defining and measuring biological activity: applying the principles of metrology. Accreditation and Quality Assurance, 12(6), 283-294. https://doi.org/10.1007/s00769-006-0254-1.
- Meza Menchaca, T., Juárez-Portilla, C., y Zepeda, R. C. (2020). Past, present, and future of molecular docking. En V. Gaitonde, P. Karmakar, y A. Trivedi (Eds.), Drug discovery and development – new advances (cap. 2). IntechOpen. https://doi.org/10.5772/intechopen.90921.
- Pavan, M., y Moro, S. (2023). Lessons Learnt from covid-19: computational strategies for facing present and future pandemics. International Journal of Molecular Sciences, 24(5), 4401. https://doi.org/10.3390/ijms24054401.
- Sarker, S. D., y Nahar, L. (2012). Natural products isolation. Methods in Molecular Biology (3a. ed., vol. 864). Humana Press. https://doi.org/10.1007/978-1-61779-624-1.
Recepción: 18/10/2022. Aprobación: 01/11/2023.